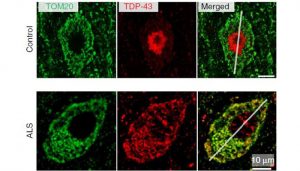
Un gran número de casos de ELA corresponde a formas hereditarias causadas por mutaciones en el gen de la superóxido dismutasa 1 (SOD1). Esta enzima disminuye los efectos del estrés oxidativo sobre las células nerviosas.
Las enfermedades de la neurona motora forman un grupo heterogéneo de síndromes que pueden ser hereditarios o no.
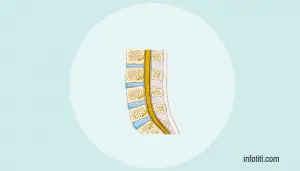
Todas tienen una base neuropatológica caracterizada por degeneración progresiva de las neuronas motoras en la corteza cerebral (motoneuronas superiores), el tronco encefálico y la médula espinal (motoneuronas inferiores).
La esclerosis lateral amiotrófica (ELA) es la enfermedad de referencia dentro de este grupo de síndromes.
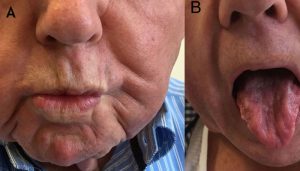
ELA se manifiesta principalmente a través de debilidad y atrofia muscular. Ambos síntomas progresan hasta el estado de parálisis y terminan afectando la autonomía motora, la comunicación oral, la deglución y la respiración.
Es más frecuente en varones y suele presentarse en individuos adultos. Además, su duración media es de solo tres años, aunque con una supervivencia de más de cinco años en el 20% de los pacientes y de más de diez en el 10%.
También te puede interesar leer: Enfermedades de la motoneurona (o lo que sucede cuando mueren las células que dirigen los movimientos)
Mutaciones en la enzima SOD1, factor de riesgo de ELA hereditaria

Las formas hereditarias son las únicas enfermedades de la neurona motora de las que se conocen las causas.
Entre el 15 y el 20% de los casos de ELA corresponde a formas hereditarias causadas por mutaciones en el gen que codifica para la enzima superóxido dismutasa 1 (SOD1).
Este hallazgo se reportó por primera vez a mediados de la década de los 90 y en poco más de 15 años ya se habían identificado más de 120 mutaciones puntuales en ese gen en pacientes con ELA hereditaria.
Como la mayoría de las mutaciones no se han logrado demostrar en múltiples familias o se relacionan con otras enfermedades neurodegenerativas, su existencia se trata como un factor de riesgo y no como una causa directa de ELA.
Dos hipótesis para explicar los efectos tóxicos de la SOD1 mutada
Las evidencias sugieren que la toxicidad mediada por la SOD1 mutada no se debe a una pérdida de su función, sino a una diversificación en las propiedades de la enzima.
Para explicar mejor este fenómeno se han propuesto dos hipótesis:
- Las SOD1 mutantes adoptan una conformación incorrecta y se agrupan en complejos de alto peso molecular que finalmente provocan la muerte de las motoneuronas.
- Las SOD1 mutantes adquieren propiedades aberrantes posibilitando la ocurrencia de reacciones que dañan sustratos críticos para la viabilidad neuronal.
Estrés oxidativo en pacientes con ELA herediaria

La SOD1 es una enzima que interviene en los mecanismos de defensa del organismo frente a la producción de radicales libres, mediante la conversión de peróxido de hidrógeno en agua.
Se ha observado que el sistema nervioso central (SNC) es más susceptible que otros tejidos al daño celular que provocan las especies reactivas del oxígeno y del nitrógeno.
Además, se ha reportado que en pacientes con ELA familiar (SOD1 mutada) aumenta el estrés oxidativo tanto en la médula espinal como en otras regiones del SNC.
Igualmente se ha comprobado la presencia de estrés oxidativo en el modelo de ratón transgénico de sobreproducción de SOD1 mutada.
Con respecto a esta última evidencia, resulta interesante cómo la administración de antioxidantes probó ser beneficiosa, ya sea retrasando el inicio de la enfermedad y/o prolongando la supervivencia de los animales.
Tras los síntomas de esta enfermedad se han descrito otros mecanismos moleculares, entre ellos: la agregación anormal de proteínas, la desorganización de los filamentos intermedios, así como alteraciones en la regulación del calcio intracelular.
Por tal razón constituye una necesidad continuar con las investigaciones, con el propósito de entender mejor sus causas y finalmente encontrar un tratamiento.
También te puede interesar leer: Tratamiento farmacoterapéutico en la esclerosis lateral amiotrófica. Estado actual y proyecciones futuras
Bibliografía
- Orient-López, R. T.-B.-E.-G. (2006). Tratamiento neurorrehabilitador de la esclerosis lateral amiotrófica. REV NEUROL 43 (9), 549-555.
Inmaculada Prior-Sánchez, A. D.-M. (2014). Gastrostomía endóscopica percutánea en esclerosis lateral amiotrófica; experiencia en un hospital de tercer nivel. Nutr Hosp. 2014;30(6):1289-1294, 1289-1294.
Prida, J. M. (2009). Esclerosis lateral amiotrófica: una actualización. Revista Mexicana de Neurociencia, 281-286.