La distrofia miotónica constituye un amplio espectro de enfermedades que afectan el metabolismo muscular, especialmente expresados por debilidad progresiva.

Se trata de una enfermedad de etiología genética expresada por trastornos musculares y visuales, aunque la gravedad de los síntomas depende mucho del tipo de enfermedad así como de la presentación clínica de cada subtipo.
Es posible identificar una distrofia miotónica tipo 1 (de Steinert) y la tipo 2 (de Ricker), que a pesar de tener mecanismos moleculares causales semejantes, también tienen considerables diferencias clínicas entre ellas. Es sobre esta particular enfermedad de lo que hablaremos a continuación.
También te puede interesar leer: Ataxia telangiectasia, cuando la inmunidad y las neuronas empiezan a fallar
¿Cuál es la causa de la distrofia miotónica?
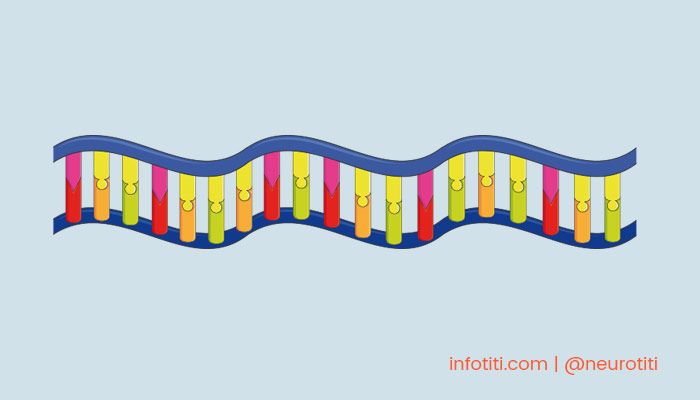
Se sabe que el defecto subyacente (por lo menos en la distrofia miotónica tipo 1) es un trastorno a nivel del material genético, aunque de manera atípica se presenta como un defecto de la integridad del ARN, un tipo de molécula muy relacionada al ADN.
El ARN es estructuralmente similar, sin embargo cumple funciones importantes especialmente durante la síntesis de proteínas en los ribosomas, unos complejos macromoleculares que existen dentro de las células y son el sitio donde realmente se sintetizan las proteínas.
Este es un proceso que requiere gran cantidad de energía en el que intervienen numerosas moléculas.
Específicamente, el daño de las distrofias se expresa como un aumento en densidad de “tripletes de citocina, tiamina y guanina (CTG)” las cuales son pequeñas moléculas llamadas bases nitrogenadas que se encuentran en el ARN (y ADN).
Todavía no se comprende exactamente cómo esto se traduce en los síntomas de la enfermedad, pero sí se sabe que entre mayor cantidad de defectos de este tipo, mayor es la intensidad de los síntomas.
La distrofia miotónica tipo 2 también tiene un defecto similar, pero involucra otro lugar del material genético y otras bases nitrogenadas.
También te puede interesar leer: Ataxia: síntomas, causas y tratamiento
¿Qué tan frecuente es?
No es una patología muy frecuente, aunque se han reportado numerosos casos en ciertas regiones sin una razón aparente. Estas zonas incluyen Suecia, Quebec y España.
Por otro lado, la información epidemiológica en América Latina es muy escasa.
¿Cuáles son los síntomas?
La distrofia miotónica tipo 1 se puede clasificar en cuatro subtipos: suave, clásica, juvenil y congénita.
La forma suave tiene muy pocos síntomas como su nombre lo indica, se caracteriza por cataratas prematuras, alopecia (calvicie) y defectos fisiológicos musculares generalmente observados mediante electromiografía.
La forma clásica inicia en la adultez temprana e incluye debilidad de las porciones distales de los miembros, cataratas, alopecia y trastornos de la conducción cardíaca.
La forma juvenil se destaca principalmente por la edad de presentación de los síntomas, apareciendo en un principio debilidad muscular en la niñez y miotonía.
También se asocia a ciertos defectos del desarrollo intelectual y social. De hecho, dado lo inespecífico de los síntomas, muchas veces el diagnóstico se hace luego de que la madre del niño haya sido diagnosticada (por la naturaleza hereditaria del trastorno).
La forma congénita es el tipo de presentación más severo de la enfermedad, manifestándose los síntomas en el período de desarrollo intrauterino.
Esto incluye disminución de los movimientos fetales, polihidramnios (es decir, un aumento de la cantidad normal de líquido amniótico generalmente medido mediante ecografía) y deformaciones congénitas.
El pronóstico es desfavorable, requiriendo estos pacientes muchas veces el ingreso a una unidad de cuidados intensivos neonatales.
Por otro lado, en el caso de la distrofia miotónica tipo 2 la edad de presentación es más tardía (adultez media) y algunos síntomas pueden pasar desapercibidos.
También te puede interesar leer: La atetosis, un ejemplo de trastorno del movimiento
¿Cuál es el diagnóstico?
Al tratarse de un trastorno genético, el diagnóstico también se establece mediante métodos moleculares destinados a evaluar el genoma.
El más conocido y práctico es la reacción en cadena de polimerasa, una técnica de biología molecular que permite la identificación de material genético anómalo proveniente de una muestra sanguínea.
La electromiografía, biopsia muscular y estudios sanguíneos que miden niveles de enzimas musculares son exámenes complementarios muy útiles.
También te puede interesar leer: Miopatía: tipos, diagnóstico y tratamiento
Fuentes y referencias:
Flores-López E, Tovilla-Ruiz C, García-Padilla E, Sandoval-Gutierrez R, Álvarez-Torrecilla L. Distrofia miotónica de Steinert: caso clínico de una familia y revisión de la bibliografía. Med In Méx 2014;30:195-203.
Crédito de imágenes: Servier Medical Art por Servier
- Historia de la medicina: el origen de la tomografía computarizada - 2020-01-19
- Historia de la medicina: Santiago Ramón y Cajal - 2020-01-19
- Apraxia de la marcha: síntomas, causas y diagnóstico - 2019-09-16